
Support for the editorial development of this report was provided by Sandoz

Alain Astier Professor of Oncology Pharmacy and Biotechnics, Henri Mondor University Hospital, Paris, France

Support for the editorial development of this report was provided by Sandoz
Alain Astier Professor of Oncology Pharmacy and Biotechnics, Henri Mondor University Hospital, Paris, France
Arnold Vulto Professor of Hospital Pharmacy & Practical Therapeutics, Erasmus University Medical Center, Rotterdam, The Netherlands

Martin Schiestl Chief Science Officer, Sandoz GmbH, Kundl, Austria

Steffen Thirstrup Adjunct Professor, Faculty of Health Sciences, School of Pharmacy, University of Copenhagen, Copenhagen, Denmark
Since the first approval in the early 1980s,1 biologics have sparked a revolution in the treatment of many diseases, including autoimmune disorders, haematological diseases and cancer.2 Many new biologic agents are approved each year, and biologics now represent more than half of the biggest selling drugs in the world.3 However, the costs of innovation are substantial, and the average cost of medicines has risen exponentially in recent decades. New cancer biologics may cost $100,000 per patient per month by 2035.4
Sustaining these costs is likely to place a considerable burden on healthcare budgets, threatening access to biologics. Indeed, inequalities in access to biologics are already evident; for example, biologics for the treatment of rheumatoid arthritis are not reimbursed at all in more than 20% of countries in Europe.5
What is a biosimilar and why are they important?
A biosimilar is a successor to a biologic medicine (known as the ‘reference medicine’) that has lost marketing exclusivity following patent expiration.6 Biosimilarity means that the biologic medicine is — as defined by the European Medicines Agency – “highly similar to another biologic medicine already marketed in the EU”.7 Due to the fact that biosimilars are made in living organisms, minor differences from the reference medicine may sometimes be present.7 However, strict controls ensure that no differences are present in attributes critical to efficacy, safety or immunogenicity, and so no clinically meaningful differences are permitted.7
In Europe, the first biosimilar was approved in 2006, and the total number of biosimilars approved for use has now increased to 23.8
Evidence suggests that use of biosimilars may positively impact on the financial sustainability of healthcare systems,9,10 addressing the unmet need for broader access to biologic therapies,10,11 without compromising patient outcomes.12 However, despite more than ten years’ experience with biosimilars in Europe, considerable scepticism and misunderstandings around biosimilars are observed amongst healthcare professionals. Indeed, surveys have shown that many physicians and pharmacists have little or no familiarity with recent biosimilar developments.12–14
Pharmacists also have concerns about biosimilar use, which may arise from lack of information on this topic. In France, for example, more than 60% of pharmacists reported limited knowledge of biosimilars, with 16% of hospital pharmacists indicating that they were ‘not at all informed’ about these medicines.15 Overall, these gaps have restricted use of biosimilars in Europe, with considerable variation in use across different countries.13,15
The barriers to biosimilar use
Pharmacists have an important role in supporting the uptake of biosimilars.2 They can act as sources of information, educate healthcare professionals and patients, and take responsibility for appropriate and safe use. However, they must be sufficiently informed and educated themselves to gain confidence in biosimilars and their use. To address some misconceptions around biosimilars and support education on this topic, a symposium, entitled ‘Hot air or hot topic? Join the biosimilar debate!’, was organised at the 22nd European Association of Hospital Pharmacists Congress (EAHP) in Cannes, on 22 March 2017. The aim was to allow interactive, in-depth, expert discussion of biosimilars for a hospital pharmacist audience and provide relevant information to address perceived barriers to biosimilar use.
To characterise the barriers, a poll of EAHP participants took place ahead of the symposium, posing the question: ‘What’s your biggest barrier to biosimilar use?’ The results (Figure 1) showed that the top four barriers (greater than 10% of participants) were immunogenicity, the concept of extrapolation for clinical indications, interpreting analytical data, and understanding the structural similarity between a biosimilar and the reference medicine.
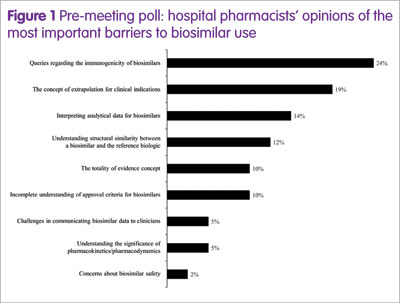
Therefore, in the symposium, four experts in hospital pharmacy, healthcare policy or biosimilar development addressed these topics in an informal debate, as shown below.
- The analytical techniques used to establish similarity Martin Schiestl (Chief Science Officer, Sandoz GmbH, Kundl, Austria)
- The regulatory pathway for biosimilars Steffen Thirstrup (Adjunct Professor, Faculty of Health Sciences, School of Pharmacy, University of Copenhagen, Copenhagen, Denmark)
- The concept of extrapolation Arnold Vulto (Professor of Hospital Pharmacy & Practical Therapeutics, Erasmus University Medical Center, Rotterdam, The Netherlands)
- Immunogenicity concerns with biosimilars Alain Astier (Professor of Oncology Pharmacy and Biotechnics, Henri Mondor University Hospital, Paris, France)
The speakers were tasked with convincing the audience that the barrier they represented should no longer be considered a barrier to biosimilar use, with audience opinion obtained via a poll.
The analytical techniques used to establish similarity
Martin Schiestl began by outlining the novel drug development paradigm for biosimilars compared with other biologics, the crux of which is “the demonstration of sameness”. Analytical data is the most important element of this process, and development begins with comprehensive characterisation of the reference medicine using an extensive array of modern analytical techniques.16 This comparative analytical data is the first step in establishing a scientific bridge to the clinical experience of the reference medicine.
Biosimilars are often perceived as somewhat different from their reference medicines. However, a prerequisite for biosimilar development is that the biosimilar and its reference medicine have an identical amino acid sequence and therefore an identical primary protein structure.17 Overall, the degree of similarity is actually extremely high (Figure 2), Martin Schiestl stated.17 Importantly, any potential difference in a quality attribute between a biosimilar and its reference medicine, such as the amount of a by-product or glycan structure, is allowed only if it is not clinically relevant.16 For approval in Europe, the US and other highly regulated markets, a biosimilar must demonstrate a match to its reference medicine in terms of safety, efficacy and quality.7
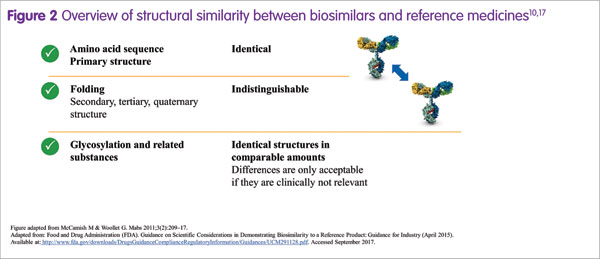
Biologics are biotechnologically engineered medicines, made by living organisms. Martin Schiestl noted that a certain degree of variability is inherent to all biologics.18 In addition, changes in the manufacturing process may be necessary periodically, such as when upscaling production, which may also contribute to variability.18,19 The result may be minor differences in some quality attributes, which potentially could impact on function. Therefore, the degree of variability is controlled within acceptable limits, and variations due to manufacturing changes are permitted by regulatory authorities only if they do not lead to any clinically meaningful differences.20 During the development of a biosimilar, extremely stringent criteria for similarity in structure and function to the reference medicine are applied, such that the biosimilar is as similar to the reference medicine as the reference medicine is to itself over time, considering the variability of the biological expression system and additional variability induced by manufacturing changes. For example, with Sandoz biosimilar rituximab (development code: GP2013), functional characterisation was done using a comprehensive range of potency bioassays. The results from these assays demonstrated that Sandoz biosimilar rituximab could not be functionally distinguished from reference rituximab.21
Due to the extensive number of quality attributes that must be assessed to demonstrate similarity, a quantitative risk assessment is used. Attributes are ranked according to their impact on safety, immunogenicity, efficacy and PK/PD, giving a criticality rating based on importance to the patient.22,23 This allows the development and production process to focus on ensuring similarity of these particularly important attributes, resulting in a precisely engineered biosimilar that matches the reference medicine in all clinically relevant attributes.
The regulatory pathway for biosimilars
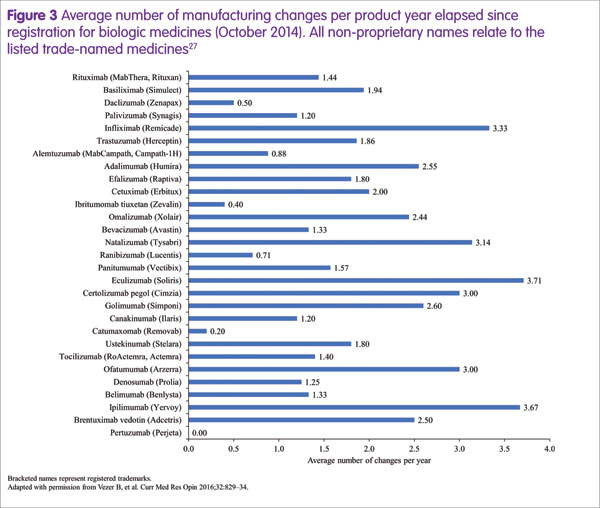
Steffen Thirstrup highlighted the approval process for a biosimilar, which is based on the highly analogous, well established approval process that is required for any biologic following manufacturing process changes.20,24–26 These changes occur frequently during the lifespan of a biologic, at an average rate of around two per year (Figure 3).27 On these occasions, comparability of pre- and post-change medicines must be demonstrated. For example, a manufacturing process change for rituximab (Rituxan®/MabThera®, Genentech/Roche) resulted in substantial changes in glycosylation that were associated with slightly increased potency of antibody-dependent cellular cytotoxicity.18 However, the regulatory authorities concluded that there was no change to the benefit–risk ratio and re-approval occurred without the need for any additional clinical data.
Biosimilars are approved on the basis of the totality of evidence concept: they must demonstrate comparability to the reference medicine in terms of analytical (physicochemical and biological characterisation), preclinical and clinical evidence.28–30 Conducting clinical trials in all potential indications does not make scientific sense, because most clinical endpoints are not sufficiently sensitive to detect potential differences between different biologics.31 Therefore, regulators suggest conducting a clinical trial using a sensitive endpoint in a sensitive indication to detect any potential clinical difference between the biosimilar and its reference medicine. Clinical data for non-studied indications can be extrapolated from the reference medicine to the biosimilar, if scientifically justified by various factors, including the degree of similarity demonstrated, detailed knowledge of the mode of action and the absence of specific safety issues.30
Steffen Thirstrup concluded by summarising the safety requirements for biologics, including biosimilars. Firstly, market exclusivity legislation ensures that there are at least ten years of market experience with the reference medicine prior to approval of a biosimilar. Also, all new biologics, including biosimilars, are subject to additional monitoring upon approval to ensure that safety is appropriately assessed. This is denoted by a black triangle on the package leaflet in Europe, indicating the precautionary principle adopted by regulators.
The concept of extrapolation
Arnold Vulto began by stating that “Extrapolation is not a fantasy or a gimmick; it’s a science”. Extrapolation is the approval of a biosimilar in a clinical indication of the reference medicine, without clinical data for the biosimilar product in that indication.30 However, extrapolation can only be granted if there is extensive evidence demonstrating that the biosimilar and reference medicine behave in the same manner in different indications. This is established by assessing the mechanism of action and target receptors across different indications, evaluating the functions of the moieties of the molecule, and assessing any actual or potential safety differences.30
Importantly, the concept of extrapolation is used constantly in all areas of medicine. For example, evidence is extrapolated from animal experiments to humans, and from clinical trials to real-world clinical practice. The scepticism to extrapolation for biosimilars is unique and not reflective of attitudes to similar cases of extrapolation. For example, both rituximab (Rituxan Hycela®/MabThera®, Genentech/Roche) and trastuzumab (Herceptin®, Roche) have undergone reformulation from intravenous to subcutaneous administration to improve convenience. The new formulations were approved by the EMA for all indications using a pathway analogous to that used for biosimilar extrapolation, and acceptance by healthcare professionals has been rapid and universal.30
Extrapolation of indications is now well-established and supported by the EMA and the International Pharmaceutical Regulators Forum.32 Furthermore, a considerable shift in attitudes to extrapolation has occurred recently among leading scientific societies, with a supportive position statements emerging from ESMO.9 Significantly, ECCO (the European Crohn’s and Colitis Organisation), which in 2013 stated that no drug should be approved in inflammatory bowel disease without a confirmatory clinical trial,33 now recommends that clinical studies in a sensitive indication can provide the basis for extrapolation.34 Arnold Vulto concluded by stating ‘If the molecule is the same, it will do the same, in all indications of the reference product.”
Immunogenicity concerns with biosimilars
Immune reactions to proteins – whether biosimilar or reference medicine – occur in two different ways, began Alain Astier. The first are neo-antigen reactions, which are immediate and acute, and generally result from non-human epitopes present as impurities.35 The second involves disruption of tolerance to a protein, which accumulates slowly after extended treatment,35 and results from binding antibodies being produced in response to protein degradation, glycosylation variants and aggregates. It is now relatively simple to characterise and remove impurities, thereby potentially reducing the risk of immunogenicity. Furthermore, any variability between a biosimilar and reference medicine is minimised via extensive comparability tests of all quality attributes, including immunogenicity.16
For protein aggregates, the level of immunogenicity is strongly dependent on the size of the aggregate, with a high level of immunogenicity for particulates of 2–10µm.36 Aggregation is a progressive process, with the initial sign being the presence of oligomers smaller than 1µm. Aggregation results in the induction of antibodies, with two potential effects: loss of efficacy caused by neutralising antibodies inactivating the therapeutic biologic, and toxicity and immunogenic reactions caused by antigenic antibodies.35 Aggregation is common to all proteins in response to mechanical stress such as shaking or stirring; shearing caused by rapid extraction from a vial via syringe; or exposure to the air.37 However, the extent of aggregation is specific to each monoclonal antibody,37,38 and is unlikely to differ between a biosimilar and its reference medicine.
“Short-term clinical trials provide limited data on immunogenicity”, stated Alain Astier, as they offer an incomplete representation of the ‘real world’. However, by the time a biosimilar is approved, the immunogenicity profile of the reference medicine is already well-known and, so far, real-world evidence studies have not revealed any differences in immunogenicity between a biosimilar and its reference medicine. Pharmacovigilance plans must be used to evaluate immunogenicity, comparing a biosimilar to its reference medicine.
Concluding remarks
To conclude his arguments, Alain Astier stated that immunogenicity is a side effect no different from any other side effect, and no evidence exists that this differs between a biosimilar and its reference medicine. Arnold Vulto repeated that “If the molecule is the same, it will do the same, in all indications of the reference product.” Steffen Thirstrup stated that the regulatory process for biosimilars is rigorous and analogous to that used for manufacturing changes of reference medicines. Finally, Martin Schiestl added that biosimilar development involves tightly controlled high-performance biotechnology, resulting in biosimilars that match the reference medicines.
Audience polling during the debate showed that immunogenicity and extrapolation remained important barriers, requiring further education. Overall, the symposium highlighted that continuing the conversation between stakeholders around biosimilars, along with continued provision of education, is vital to support informed decision-making and overcome the barriers to biosimilar use.
References
1 Quianzon CC, Cheikh I. History of insulin. J Community Hosp Intern Med Perspect 2012; 2. doi: 10.3402/jchimp.v2i2.18701.
2 Stevenson JG et al. Biosimilars. Ann Pharmacother 2017 Feb 1:1060028017690743. doi: 10.1177/ 1060028017690743.
3 PharmaCompass. Top drugs by sales revenue in 2015: Who sold the biggest blockbuster drugs?
www.pharmacompass.com/radio-compass-blog/top-drugs-by-sales-revenue-in-2015-who-sold-the-biggest-blockbuster-drugs (accessed September 2017).
4 Cornes P. Introduction: Finpro genomics collaboration event, London, November 2016. www.finpro.fi/documents/1431875/0/Cornes+Introduction+ Finpro (accessed September 2017).
5 Putrik P et al. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis 2014;73:198–206.
6 European Medicines Agency. Biosimilar medicines. www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/document_listing/document_listing_000318.jsp (accessed September 2017).
7 European Medicines Agency. Biosimilars in the EU, Information guide for healthcare professionals. www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf (accessed September 2017).
8 European Medicines Agency. European public assessment reports. Browse by type. Biosimilars. www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124 (accessed September 2017).
9 Tabernero J et al. Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open 2016;1:e000142. doi:10.1136/esmoopen-2016-000142.
10 McCamish M et al. Worldwide experience with biosimilar development. mAbs 2011;3:209–17.
11 McCamish M et al. Biosimilars: biologics that meet patients’ needs and healthcare economics. Am J Manag Care 2016;22:S439–442.
12 Zelenetz AD. Biosimilars in oncology. Oncol Hematol Rev 2016;12:22–8.
13 Pasina L et al. A survey among hospital specialists and pharmacists about biosimilars. Eur J Intern Med 2016 Nov;35:e31–33.
14 Cohen H et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther 2017;33:2160–72.
15 Beck M et al. Knowledge, behaviors and practices of community and hospital pharmacists towards biosimilar medicines: Results of a French web-based survey. MAbs 2017;9:383–90.
16 Windisch J. EGA’s perspective on the draft quality guideline. www.ema.europa.eu/docs/en_GB/document_library/Presentation/2013/11/WC500154191.pdf (accessed September 2017).
17 Food and Drug Administration (FDA). Scientific considerations in demonstrating biosimilarity to a reference product: Guidance for Industry (April 2015). www.fda.gov/downloads/DrugsGuidanceCompliance RegulatoryInformation/Guidances/UCM291128.pdf (accessed September 2017).
18 Schiestl M et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol 2011;29:310–12.
19 Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis 2013;72:315–18.
20 European Medicines Agency (EMA). ICH Q5E: Comparability of Biotechnological/Biological Products. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002805.pdf (accessed September 2017).
21 Visser J et al. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013;27:495–507.
22 FDA Arthritis Advisory Committee, 13 July 2016. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM513088.pdf (accessed September 2017).
23 ICH Q8(R2) Guideline. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf (accessed September 2017).
24 European Medicines Agency (EMA). Guideline on comparability of biotechnology-derived medicinal products after a change in the manufacturing process: non-clinical and clinical issues. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003935.pdf (accessed September 2017).
25 European Medicines Agency (EMA). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/ 2013/06/WC500144124.pdf (accessed September 2017).
26 European Medicines Agency (EMA). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (rev 1) www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf (accessed September 2017).
27 Vezer B, et al. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin 2016; 32:829–34.
28 Ebbers HC. Biosimilars: in support of extrapolation of indications. J Crohns Colitis 2014;8:431–5.
29 Weise M et al. Biosimilars: what clinicians should know. Blood 2012;120:5111–17.
30 Weise M et al. Biosimilars: the science of extrapolation. Blood 2014;124:3191–6.
31 Singh JA et al. Cochrane Database Syst Rev
2009; Oct 7:CD007848. doi: 10.1002/14651858.CD007848.pub2.
32 International Pharmaceutical Regulators Forum. IPRF biosimilar working group draft reflection paper on extrapolation of indications in authorization of biosimilar products. 25 July 2016. www.i-p-r-f.org/files/1614/7315/4963/160725_IPRF_BWG_Reflection_paper_final_draftver_1.3_IPRF_MC_Circulation.pdf (accessed September 2017).
33 Danese S et al. ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohns Colitis 2013;7:586–9. doi: 10.1016/j.crohns.2013.03.011.
34 Danese S et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease-an update. J Crohns Colitis 2017;11:26–34.
35 Ratanji K et al. Immunogenicity of therapeutic proteins: Influence of aggregation. J Immunotoxicol 2014;11:99–109.
36 Wang W et al. Immunogenicity of protein aggregates–concerns and realities. Int J Pharm 2012;431:1–11.
37 Lahlou A et al. Mechanically-induced aggregation of the monoclonal antibody cetuximab. Ann Pharm Fr 2009; 67:340–52.
38 Paul M et al. Long-term stability of bevacizumab repackaged in 1mL polypropylene syringes for intravitreal administration. Ann Pharm Fr 2012; 70:139–54.